Quick Start
1. Input Parameters
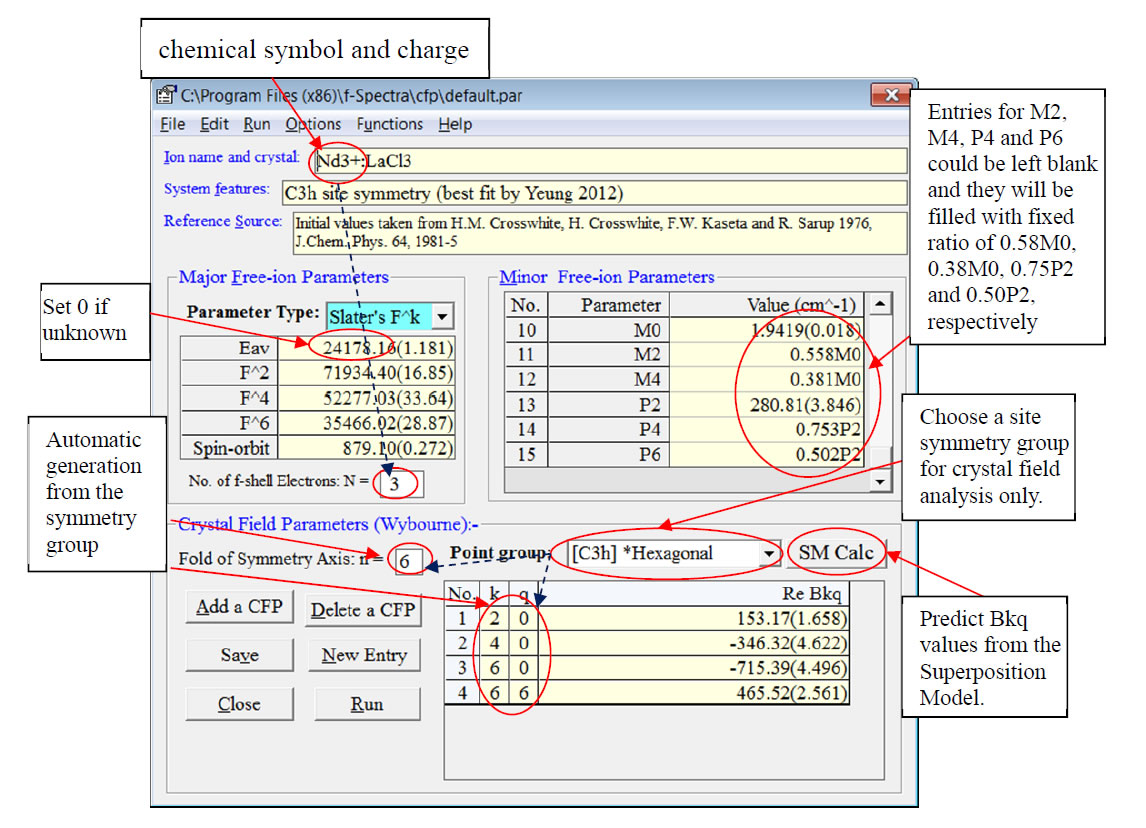
Figure 1: Window for input of free-ion and crystal field parametersIf you cannot find the above window, you may click the menu Run->Input Parameters in the main window with the caption “f-Spectra (f-shell Spectroscopic Analysis Package)”.
2. Calculation of Crystal Field Energies
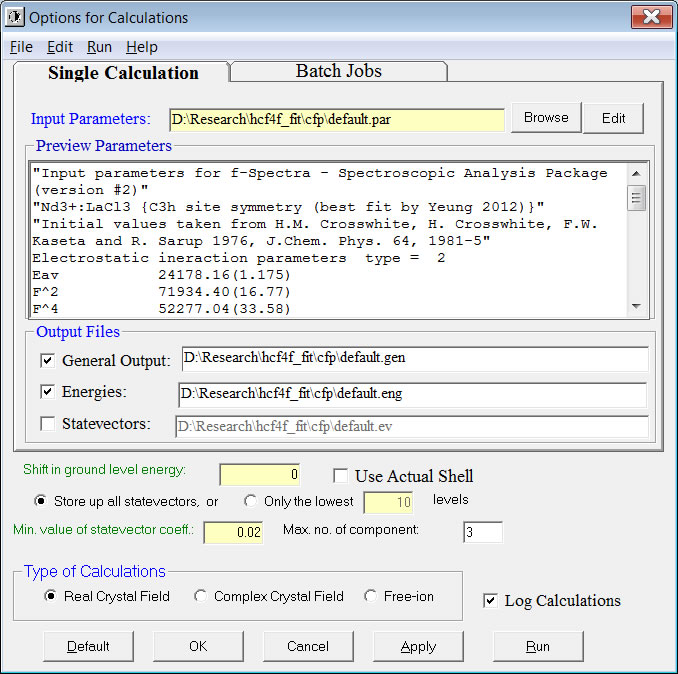
To calculate the energy levels using the given parameters, choose Run->Calculations with Options->Real/Complex Crystal Field from the menu. The following “Options for Calculations” window will be displayed.
If the default settings are OK, you should click the Run button to start the calculation which will usually be completed within a minute for a system with less than 4 or more than 10 f-shell electrons. For those systems with 4 to 10 f-shell electrons at low-symmetry sites, it may take several minutes up to around an hour, depending on the speed of your computing machine.
Three plain texts will be generated and shown with the following file name extension (with filename taken from that of the *.par file):
- *.eng for the label 2S+1LJ, crystal quantum number mu, irreducible representation IR, energy, Zeeman splitting and s’ (with second order correction), degeneracy and 3 largest statevector components of each crystal field level.
- *.ev for the coefficients of every statevector (quantum numbers for each statevector component are given at the beginning of the file).
- *.gen for general information of the fit or calculation, including input parameters, output energies and statevectors etc.
3. Fitting to Observed Crystal Field Energy Levels
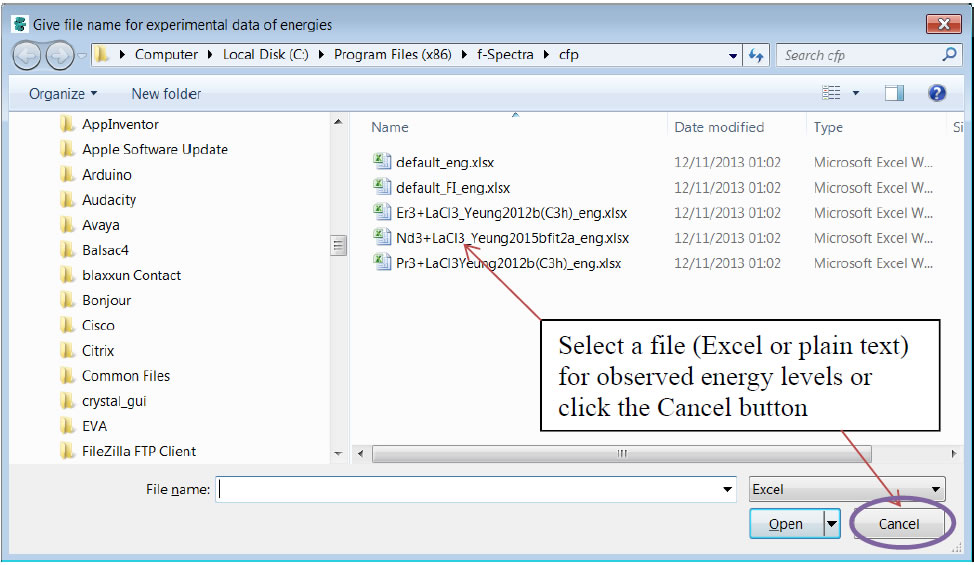
Figure 2: window “Get file of experimental data for energies”
- Then, the following Fitting window will be displayed to show the input parameter values and the observed energies.
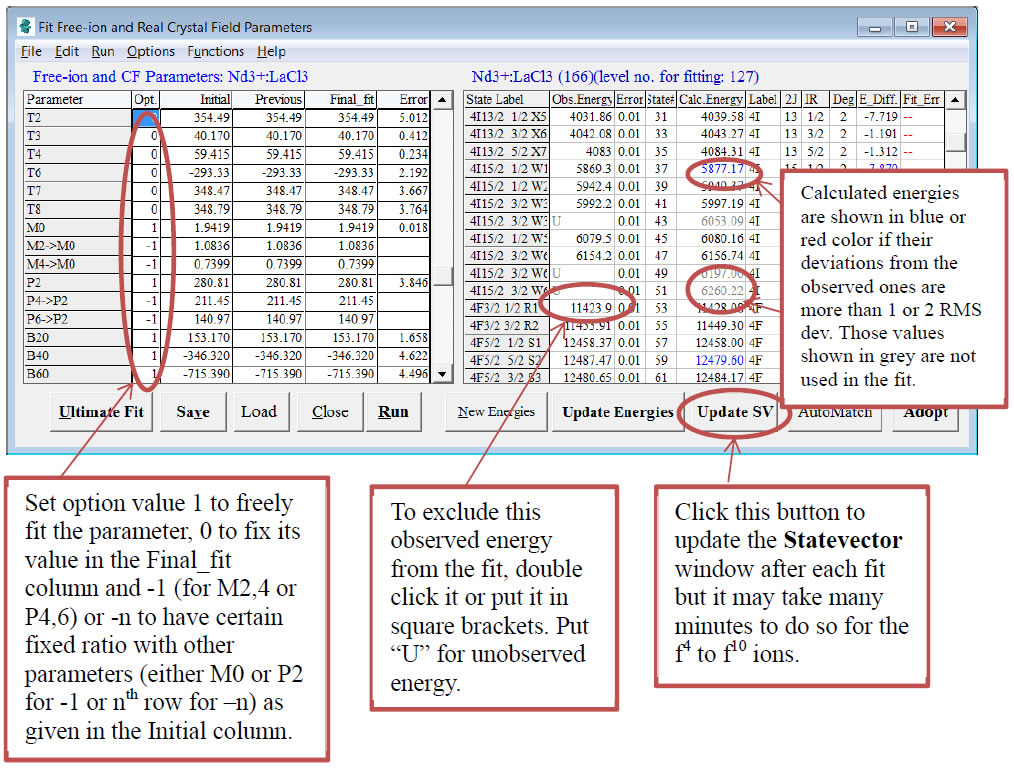
Figure 3: Fit Window to control the fit processes and to show the results of fits or calculations
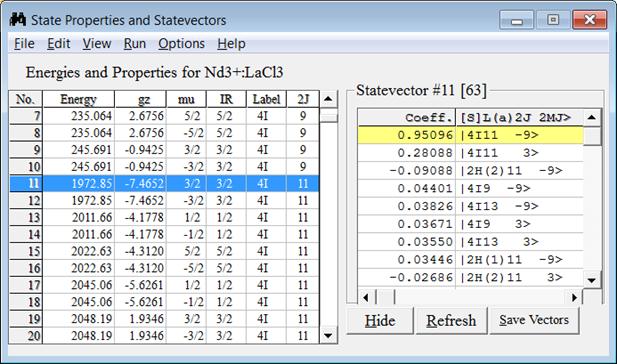
Figure 4: State Properties and Statevectors window to show the energies, spectroscopic properties (like the g-factor for Zeeman splitting, mu for crystal quantum number, IR for irreducible representation, (2S+1)L label and J value) and statevector components.
- In the LHS table of the Fit Window, set option value in the Opt. column as follows:
- 1 to freely fit the parameter,
- 0 to fix its value in the “Final_fit” column and
- -1 (for M2,4 or P4,6) to have certain fixed ratio with other parameters (either M0 or P2 for -1 or a particular parameter in the nth row for –n) as given in the Initial column. The linked parameter will be indicated with the “->” symbol.
To carry out the fit, click the [Run] button or select Run->Free-ion and CF parameters fit. After each round of fit, you should click the [Update Energies] or [Update SV] button to update the calculated values of the energies or statevectors (including update of energies). The latter process is required before you carry out any refit.
[Previous: Introduction and Installation]|[Table of Contents]|[Next: Advanced Topics]
Copyright (C) 2016. YEUNG Yau Yuen. All Rights Reserved.